Methods for Inference of Demography and Selection
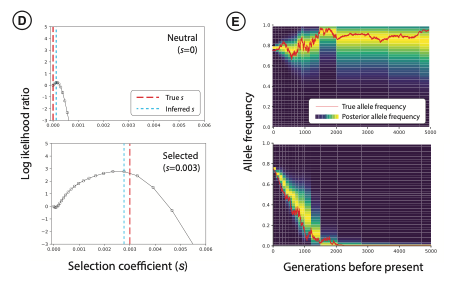
A core line of research in the group is the development of computational methods for inference of complex demographic models. For example, we have previously
developed Markov Chain Monte Carlo (MCMC) methods for joint estimation of population divergence times,
changes in population size, gene-flow between populations,
and other parameters together with Jody Hey at Temple University and implemented in the
IM programs. We have also developed a number of methods
for detecting natural selection from DNA sequence data, including the methods implemented in
SweepFinder2,
VolcanoFinder and
Ohana.
We are currently focusing on developing rigorous methods for using Ancestral Recombination Graphs for
inferences of demography and selection, so far resulting in the methods implemented in
CLUES,
CLUES2,
PALM,
SINGER, and
POLEGON.
Example Papers
- Robust and accurate Bayesian inference of genome-wide genealogies for hundreds of genomes
- A general framework for branch length estimation in ancestral recombination graphs
- Disentangling selection on genetically correlated polygenic traits via whole-genome genealogies
- An approximate full-likelihood method for inferring selection and allele frequency trajectories from DNA sequence data
- Joint Estimation of Pedigrees and Effective Population Size Using Markov Chain Monte Carlo
- Estimating the timing of multiple admixture pulses during local ancestry inference
Bioinformatics for Next Generation Sequencing Data
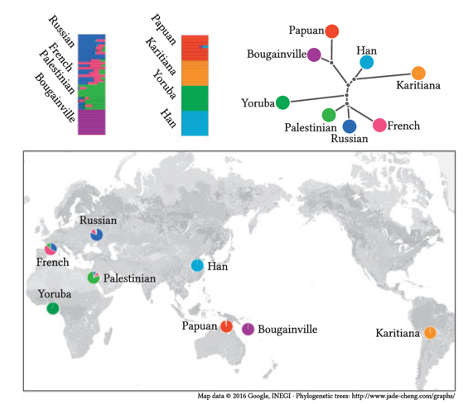
Over the past 15 years we have developed a number of statistical and computational
tools used in analyses and interpretations of NGS data. Many of the methods are made
available to other researchers in the
ANGSD package.
These methods are designed for non-model organisms for which imputation-based genotype calling is unreliable
or impossible. Instead, we model the uncertainty in the genotype calls using genotype likelihoods and
incorporate genotype uncertainty in downstream analyses. This approach allows researchers to leverage
inexpensive low-coverage sequencing data for advanced population genetic analyses.
Example Papers
- Fast admixture analysis and population tree estimation for SNP and NGS data
- Estimating IBD tracts from low coverage NGS data
- ANGSD: Analysis of Next Generation Sequencing Data
Human Genetic Diversity and Disease
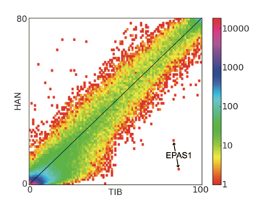
Most human genetic studies have focused on European and other highly represented populations.
However, much of human genetic and phenotypic variation is not represented in this group.
Focusing primarily on Europeans limits our ability to provide equal medical treatment for all
population groups. Furthermore, understudied populations harbor variants not commonly found in
Europeans that can help provide a better understanding of disease etiology for the benefit of all.
My group has specialized in analyzing genetic variation in understudied groups, including Inuit
from Greenland, Tibetans, Aboriginal Australians, Native Americans, and other groups. For example,
we have shown that Tibetans harbor unique genetic variants that modulate the regulation of erythropoiesis
to avoid mis-regulation in high altitude. We have also shown that these variants were transferred to
Tibetans by interbreeding with extinct hominids called ‘Denisovans’.
In studies of Aboriginal Australians we have identified unique genetic adaptations associated with
thyroid function, likely related to thermal regulation. In Inuit from Greenland, we have identified
mutations that alter the regulation of fatty acid desaturase (FADS) genes, thereby decreasing the endogenous
synthesis of certain poly-unsaturated fatty acids (PUFAs) to compensate for a high dietary intake of these fatty acids.
Example Papers
- Human Disease Variation in the Light of Population Genomics
- Human adaption to extreme environmental conditions. Current opinion in genetics & development
- Physiological and Genetic Adaptations to Diving in Sea Nomads
- Natural selection on genes related to cardiovascular health in high-altitude adapted Andeans
- Tracing the peopling of the world through genomics
Analyses of Ancient DNA
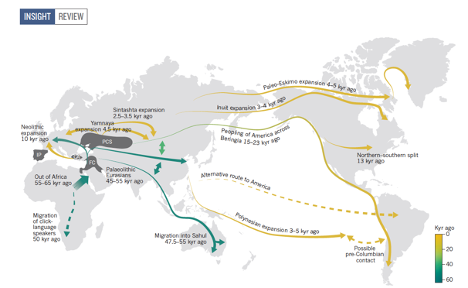
We work together with Eske Wilerslev and other researchers at
the
Lundbeck Foundation Center for GeoGenetics at the University of Copenhagen
on analyzing ancient DNA. The questions are broad but include patterns of origins and dispersal of
humans and our adaptation to local environments. We have particularly worked on the peopling of the
Americas and on European patterns of migration and dispersal from the Mesolithic to early Middle Ages.
Example Papers
- Population genomics of the Viking world
- The population history of northeastern Siberia since the Pleistocene
- Early human dispersals within the Americas
- 137 ancient human genomes from across the Eurasian steppes
- The first horse herders and the impact of early Bronze Age steppe expansions into Asia
- Terminal Pleistocene Alaskan genome reveals first founding population of Native Americans
Viral Evolution
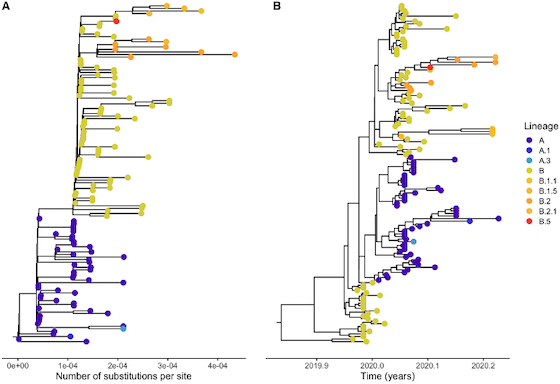
We have on and off been working on viral evolution for the past several decades.
After the emergence of SARS-CoV-2 we have focused renewed attention on this area to understand
the evolution of the virus. We expect viral evolution to continue to be an important area of
focus in the lab.
Example Papers
- Synonymous mutations and the molecular evolution of SARS-Cov-2 origins
- Assessing uncertainty in the rooting of the SARS-CoV-2 phylogeny
- A Bayesian framework for inferring the influence of sequence context on point mutations
- Passage adaptation correlates with the reduced efficacy of the influenza vaccine
- The evolutionary pathway to virulence of an RNA Virus
Herpetogenomics

We love reptiles and amphibians and maintain several research projects on their genomics
and evolutionary genetics. We are particularly fond of poison frogs, such as the lovely
strawberry frog (
Oophaga pumilio) shown in the picture. In several research projects, in collaboration
with
Kyle Summers and
Corinne Richards-Zawacki, we are trying to elucidate the
genetic basis of color variation in poison frogs. We also have ongoing research on the genomics
of frogs living in high altitude. Finally, we work on mapping of phenotypes and behavior in
the side-blotched lizard.
Example Papers
- The genetics, evolution, and maintenance of a biological rock-paper-scissors game
- Selection-driven color variation in the aposematic strawberry poison frog, Oophaga pumilio
- Divergence, gene flow, and the origin of leapfrog geographic distributions: The history of colour pattern variation in Phyllobates poison‐dart frogs
- Genomic takeover by transposable elements in the strawberry poison frog
- The genetic basis of adaptation following plastic changes in coloration in a novel environment
Phylogenetics and Comparative Analyses
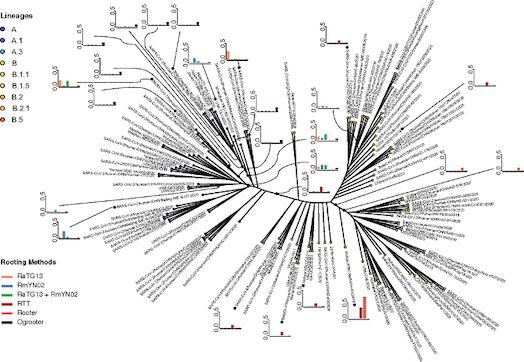
We have worked extensively on methods for phylogenetic analyses, particularly for using
phylogentic trees for inferences in molecular evolution. Many of the methods we have helped
develop are implemented in
the
PAML package
by
Ziheng Yang. We have also worked on modeling expression
level evolution along the lineages of a phylogeny and on various statistical methods for
imputation using phylogenies. We are currently working on using large phylogenies
(>100k leaf nodes) for inference of evolutionary processes.
In collaboration with Chao Zhang and Siavash Mirarab, we have also developed new methods for species tree estimation from genomic data, implemented in the
ASTER software package.
Example Papers
- ASTER: A Package for Large-Scale Phylogenomic Reconstructions
- CASTER: Direct species tree inference from whole-genome alignments
- Assessing uncertainty in the rooting of the SARS-CoV-2 phylogeny
- Phenotypic convergence is not mirrored at the protein level in a lizard adaptive radiation
- Phylogenetic ANOVA: The Expression Variance and Evolution Model for Quantitative Trait Evolution
Methods for Environmental DNA Analyses
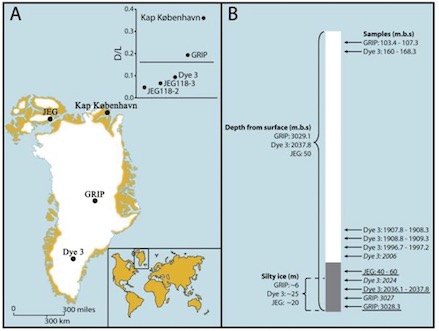
We have on and off been working on DNA barcoding and analyses of environmental DNA over the past 15 years.
This is an area of very active research in the lab at the moment. We focus on developing better methods
for species assignment and on developing methods for leveraging eDNA for population genetic analyses.
Example Papers
- Fast phylogenetic DNA barcoding
- Statistical Assignment of DNA Sequences Using Bayesian Phylogenetics
- Ancient Biomolecules from Deep Ice Cores Reveal a Forested Southern Greenland
- Ancient DNA Chronology within Sediment Deposits: Are Paleobiological Reconstructions Possible and is DNA Leaching a Factor?
Evolutionary Genomics of Domesticated Plants and Animals
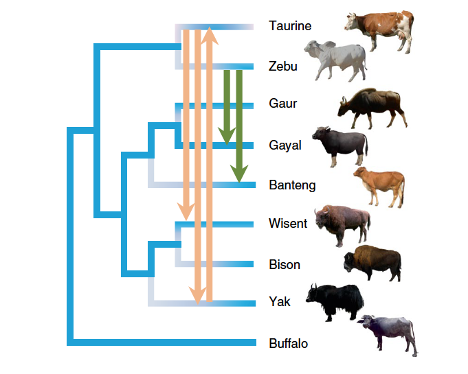
We have worked on multiple research aimed at understanding evolutionary processes during domestication,
particularly the way domestication selection has affected genetic variation and the consequences
of admixture with wild species. We are currently working particularly on rice and on betta fish.
Example Papers
- Ancient hybridization with an unknown population facilitated high-altitude adaptation of canids
- Pervasive introgression facilitated domestication and adaptation in the Bos species complex
- Asian wild rice is a hybrid swarm with extensive gene flow and feralization from domesticated rice
- The power of inbreeding: NGS based GWAS of rice reveals convergent evolution during rice domestication
Cancer Evolution and Cell Lineage Trees
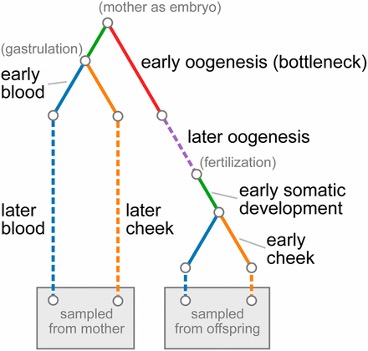
Much of our previous work has focused on developing computational methods to elucidate
evolutionary changes in DNA or in phenotypes such as expression levels, along the branches
of evolutionary trees. We are now using similar computational techniques to elucidate
processes along the branches of cell lineage trees within an individual. The process of
differentiation of cells during ontogeny/morphogenesis can be described by a tree, much
like an evolutionary tree. Similarly, the process of mutation and cell differentiation
driving carcinogenesis can be described as an evolutionary process acting within an individual
in which the unit of evolution is a cell instead of an individual. A new line of research in
the lab is the modeling and analysis of somatic mutations.
We have worked with
Kateryna Markova on
analyzing mitochondrial heteroplasmies and on developing probabilistic models of the spread of
somatic mutations during ontogenesis. These models allow us to take advantage of large
NGS data sets to test hypotheses regarding differential proliferation of mutations in different
tissues and on natural selection acting on the population of cells within an individual.
We are also working on modeling single-cell sequencing data from tumors to provide better
computational methods for analyzing tumor evolution.
Example Papers
- Pronounced somatic bottleneck in mitochondrial DNA of human hair
- Bottleneck and selection in the germline and maternal age influence transmission of mitochondrial DNA in human pedigrees
- A population phylogenetic view of mitochondrial heteroplasmy